[Förtydligande: Det som skrivits i denna artikel skall och får inte uppfattas som ett medicinskt råd i enskilda sjukdomsfall utan skall betraktas som akademiska spekulationer med intressanta nya infallsvinklar för de läkare, sjuksköterskor, forskare och lekmän som är intresserade av ämnet. Undertecknad av denna artikel, docent Sture Blomberg, är inte onkolog och har ingen egen erfarenhet av behandling av patienter med cancer och varken vill, bör, kan eller får ge råd, som går emot behandlande läkares råd.]
Indiska forskare har ägnat flera decennier åt studier kring fenbendazol och cancer. Jag kommer nu att gå igenom några av deras fynd, såsom de beskrivits i en lång och omfattande artikel från 2018: [Nilambra Dogra et al.”Fenbendazole acts as a moderate microtubule destabilizing agent and causes cancer cell death by modulating multiple cellular pathways.” Sci Rep. 2018; 8: 11926. Published online 2018 Aug 9.].
I stort sett pekar de på tre effekter, som var för sig är tillräckliga för att få cancercellen att begå programmerat självmord (apoptos), men som också samverkar med varandra och kan ge de dramatiska effekter som beskrivits av Joe Tippens. Dessa effekter är:
1. Fenbendazol bryter ner och förstör de rör (tub=rör) i cancercellen (mikrotubuli) som bl. a. behövs för att cancercellen skall kunna dela sig och växa till en cancersvulst.
2. Fenbendazol stimulerar proteinet p53, som får cancercellen att begå programmerat självmord (apoptos).
3. Fenbendazol förhindrar på flera olika sätt att cancercellen kan livnära sig på socker som är ett absolut behov hos cancercellen, varför cancercellen dör av svält.
: Fenbendazols tre verkningsmekanismerBakgrund till mekanism 1.
En cell kan liknas vid ett litet samhälle med bibliotek och kopiator i centrum (cellkärnan), och utanför detta proteinfabriker, truckar som hämtar delar från lagret, energiverk, postverk som stämplar breven med rätt adress, vägnät, sopstationer, samt låsta portar runt samhället som öppnas med kodsystem inte alls olikt våra streckkoder, telefoner. [DNA, messenger-RNA, ribosomer, transfer-RNA, aminosyror, mitokondrier, Golgiapparat, endoplasmatiskt retikulum, lysosomer, receptorer, signalsystem].
Men för att inte hela cellen skall kollapsa av trycket från omgivningen så har den också vissa stödjestrukturer ungefär som att ett hus måste ha lodräta stödjepelare för att inte kollapsa. Det finns tre olika sådana, bl. a. något som kallas mikrotubuli. Dessa byggs upp av två sorters proteiner som kallas ⍺- och 𝛃-tubulin, som tillsammans skapar rörformiga strukturer för bättre hållfasthet (tub=rör, tänk på cykelramar, som är rörformiga). Dessa rörformiga strukturer, mikrotubuli, förkortas och förlängs mycket snabbt vilket har en avgörande betydelse bl. a. då en cell skall dela sig och bli till två celler. Det finns på Internet flera för lekmän lättfattliga och instruktiva YouTube-föreläsningar om hur mitosen går till, som kan rekommenderas, bl. a. denna: [https://www.youtube.com/watch?v=C6nIGaK5900&t=515s] Det framgår i denna video hur olika mikrotubuli under själva delningsprocessen fäster med ena ändan på vardera sidan av cellväggen (som två poler) och med sin andra ända fäster på den kromosomdublett som skall till vardera av de nu två nya cellerna. Och delningsprocessen tillgår så att när mikrotubuli förkortas så ”drar” den sin kromosomdel till sin nya cell. Om mikrotubuli är skadade på något sätt så kan inte cellen dela sig och bli två, vilket således kan bli ett kritiskt moment när det handlar om cancerceller.
Fenbendazol
Sådana substanser som Fenbendazol har rapporterats hämma uppbyggnaden av mikrotubuli (polymerisationen av tubulin) och därför störa mikrotubulis funktion i parasitceller [Laclette JP, Guerra G, Zetina C. Inhibition of tubulin polymerization by mebendazole. Biochem Biophys Res Commun. 1980;92:417–423.] [Gull K, Dawson PJ, Davis C, Byard EH. Microtubules as target organelles for benzimidazole anthelmintic chemotherapy. Biochem Soc Trans. 1987;15:59–60. ]. I denna studie ville man se vilken effekt Fenbendazol har på humana cancerceller jämfört med friska däggdjursceller.
a. Fenbendazol destabiliserar tubulinnätverket i cancerceller
Humana icke-småcelliga lungcancerceller från en speciell patient (denna cellinje kallas A549) odlades och behandlades med 1 µM Fenbendazol (FZ) under 24 timmar. Organisationen av mikrotubuli efter FZ-behandling visualiserades i mikroskop genom immunfluorescens med användning av anti-⍺-tubulinantikropp som ger grön lysande färg. Cellkärnorna motfärgades röda med propidiumjodid.
När man behandlade mänskliga lungcancerceller med Fenbendazol så fick man en kraftig förändring av mikrotubulis nätverk i cancercellen. Man ser tydligt hur nätverket runt kärnan har blivit mycket glesare (se fig. 1) och därför kan förmodas få svårigheter att genomföra celldelningen.
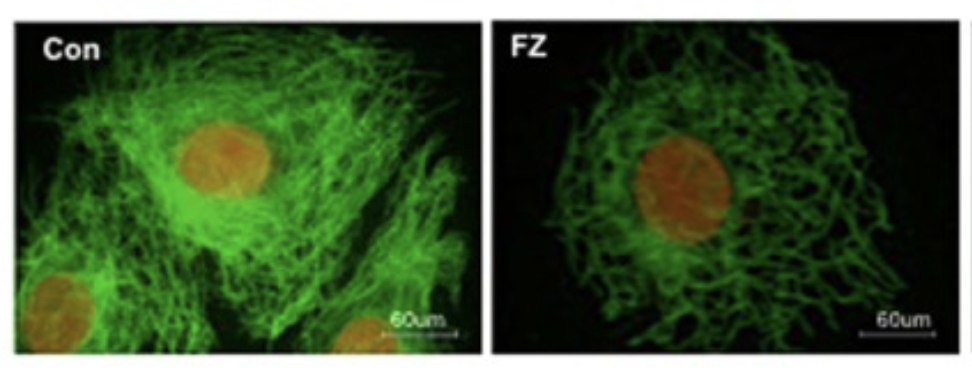
b. Fenbendazol påverkar inte mikrotubuli i friska celler
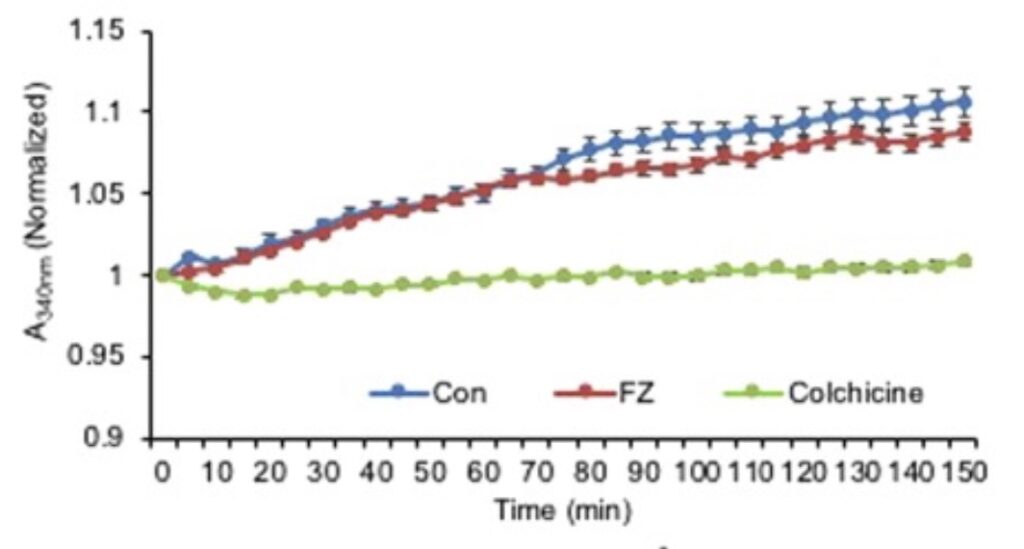
När man behandlade friska däggdjursceller med Fenbendazol så fick man bara en minimal påverkan på uppbyggnaden (polymeriseringen) av mikrotubuli (se fig. 2). Som en jämförelse har man också behandlat med giktmedlet colchicin, som sedan lång tid tillbaka är känd för att förhindra att cellen kan dela sig och därför används i denna typ av experiment.
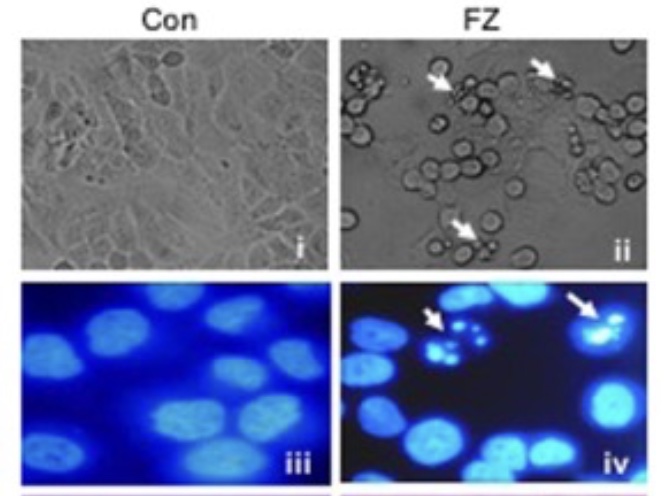
c. Fenbendazols påverkan på cancerceller leder till apoptos (programmerat självmord).
Eftersom hämning av mikrotubulis uppbyggnad (polymerisationen av tubulin) blockerar cellens vidare utveckling och gör att cellen inte kan dela sig och föröka sig, undersöktes effekten av 1µMol Fenbendazol på cellcykelns progression hos mänskliga icke-småcelliga cancerceller (A460).
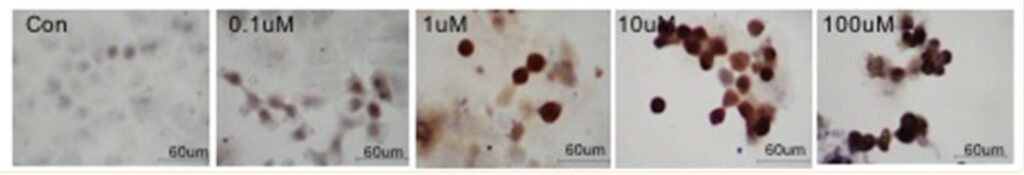
Det bekräftades att Fenbendazol orsakade stopp i själva delningsfasen av cellens utveckling till två nya celler. Efter bara 32 timmars behandling hade 30% av cancercellerna redan genomgått apoptos (= programmerat självmord). Genom att också efter 40 och 48 timmar mäta ett annat viktigt ämne (cyklin B1) kunde man visa att detta programmerade självmord (apoptos) fortsatte och drog in ännu flera cancerceller. Man kunde visa att Fenbendazol tvingade celler att stanna upp och ansamlas i den fas som skulle leda till celldelning (mitos), men att dessa celler inte kunde gå vidare till celldelning, vilket ledde till en dramatisk ökning av apoptos.
Figur 3 visar apoptotiska celler efter 24 timmars behandling med Fenbendazol. Sammantaget tyder dessa data på att Fenbendazol orsakar stopp i cellcykeln och mitotisk celldöd. Och detta kan stämma väl med att Fenbendazol hämmar tillväxten av mikrotubuli och förhindrar att mikrotubuli kan dra kromosomer till varsin hälft av cellen under mitosen.
Bakgrund till mekanism 2:
p53 i cellkärnan – skyddar mot cancer
I kroppen bildas det hela tiden skadade celler som kan utvecklas till cancer. Att dessa inte utvecklas till stora tumörer beror på att proteinet p53 ser till att skadan i DNA:t antingen: a. repareras, b. den skadade cellen förhindras att dela sig och på så sätt inte kan utvecklas till en större tumörmassa eller c. att den genomgår programmerat självmord (apoptos). Proteinet p53 har studerats i 50 år och är kanske det mest studerade av alla proteiner. Man har beskrivit p53:s roll som en ”scanning” av hela cellens DNA. Resultatet av denna scanning ger sedan upphov till syntes av de olika proteiner som krävs för en a. DNA-reparation, att b. förhindra celldelning eller c. orsaka programmerat självmord (apoptos). För att kunna genomföra en sådan ”scanning” av DNA:t krävs emellertid fyra helt identiska och icke muterade p53 proteiner som samverkar. Om en eller flera av dessa p53 har muterat så kan inte reparationen utföras. Det är därför inte förvånande att mutationer i den gen som kodar för detta protein p53 är orsaken till 50% av all cancer hos människa. De andra 50% beror på mutationer i andra gener, som är delaktiga i regleringen av p53:s aktivitet.
p53 också utanför cellkärnan – skyddar mot cancer
Men p53 finns inte bara i cellkärnan utan också i en separat pool i cellens cytosol utanför cellkärnan. Detta p53 hålls emellertid inaktivt genom att det binds till ett annat protein MDM2, vilket markerar att cellen är frisk och varken behöver repareras eller dödas. Detta p53 är således överflödigt varför merparten bryts ner och förstörs genom en speciell mekanism i cytosolen, proteasomen.
Så fort en cells DNA är skadat eller allvarligt förändrat så släpper emellertid bindningen till MDM2. Att bindningen till MDM2 upphör markerar att denna cell är sjuk och att p53 istället skall aktiveras för sin uppgift att utföra programmerat självmord (apoptos). Det bildas då mycket obundet p53 i cytosolen, vilket tar sig in i både cellkärnan och till mitokondriets yttre membran, varvid cancercellens programmerade självmord (apoptosen) kan utlösas både från cellkärna och mitokondrie. (Marchenko et al, 2000; Sansome et al, 2001; Dumont et al, 2003; Gilman et al, 2003; Mihara et al, 2003; Bonini et al, 2004; Dagher, 2004; Erster et al, 2004; Leu et al, 2004; Nemajerova et al, 2005b; Endo et al, 2006; Strom et al, 2006).
Cytokrom C
Cytokrom C, som är beläget i elektrontransportkedjan i mitokondriens inre membran, är en absolut nödvändig förutsättning för cellens produktion av energi. Men med ökad mängd och ökad aktivitet av p53, som blir fallet då cellens DNA skadats eller muterat, kommer p53 att förflyttas till mitokondriens yttre membran och göra detta genomsläppligt för olika molekyler, bl. a. Cytokrom C. När Cytokrom C från elektrontransport-kedjan i mitokondriens inre membran emellertid läcker ut genom mitokondriens nu genomsläppliga yttre membran till cellens cytosol så aktiverar detta en kaskad av enzymer, caspaces, vilka bryter ner cellens proteiner och dödar cancercellen. (https://www.youtube.com/watch?v=paw2SUY22_s)
Fenbendazol framkallar apoptos också via p53
”Vildtyp p53”
”Vildtyp p53” kallas den naturliga och icke muterade formen av p53 medan den muterade formen kallas ”p53-mutans”. Med ”p53-noll” avses en så svår mutation att den inte kan färgas och därför inte heller kan upptäckas. Två av de cellinjer som använts i dessa försök, H460 och A549, kommer från patienter med icke-småcellig lungcancer där dock deras p53 har varit ”Vildtyp p53”, dvs. icke muterat medan två andra cellinjer, H522 och H1299, var av de senare typerna. Detta är viktigt att hålla i minnet när vi går in på nästkommande försök.
Tumörcellinjer med ”vildtyp” p53 går lättare i apoptos (programmerat självmord) under inverkan av Fenbendazol
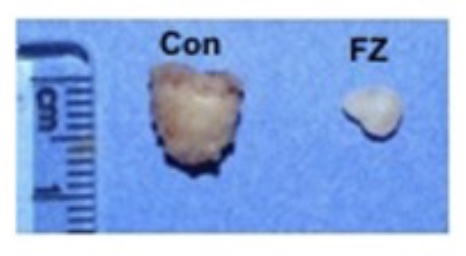
Tumörcellinjer med vildtyp p53 verkade vara mycket känsliga för Fenbendazol jämfört med celler med p53 som hade muterat. Behandling med 0,1-5 µM Fenbendazol av två olika humana cellinjer av icke-småcellig lungcancer, H460 och A549, reducerade nämligen tumörcellernas tillväxt med upptill 70-80%. (se Fig. 4).
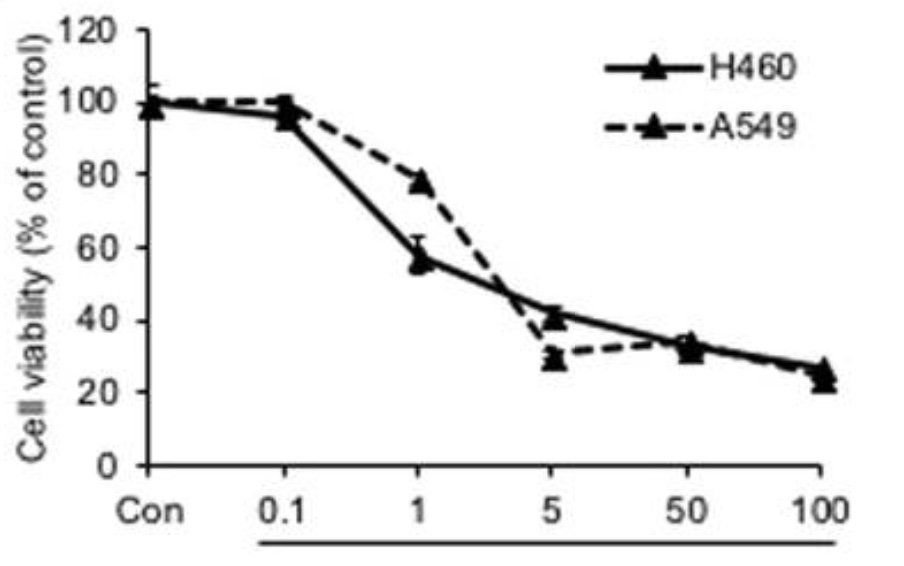
Fig. 4. Humana lungcancerceller av typ H460 eller A549 ströks ut på vävnadsodlingsplattor. Celler lämnades obehandlade eller behandlades med olika doser av Fenbendazol (0,1 – 100 µM). Efter 48 timmar analyserade man hur många procent av cellerna som levde efter ingen behandling alls (kontroll) eller efter behandling med olika koncentrationer av Fenbendazol. Man kan se att 1µM i den ena celllinjen är tillräckligt för att döda 40-50%. I den andra cellinjen dör c:a 75% av 5µM. Om man därutöver ökar dosen 10- eller 20-faldigt så har man emellertid ingen ytterligare vinst. (Bilden från Nilambra Dogra et al. ovan)
När man tillsatte vildtyp p53 till de cellinjer som inte hade detta (H1299) utan var muterade (”p53-noll”) ökade apoptosen signifikant även här och man kunde påvisa 50% flera döda cancerceller. (Figuren visas inte här med kan återfinnas i originalartikeln som figur 5c.)
Friska celler påverkas inte
Fenbendazol visade däremot mindre toxicitet mot celler odlade från frisk lungvävnad på råtta jämfört med lungcancerceller från människa. Detta visar att Fenbendazol i närvaro av vildtyp p53 framkallar ökad apoptos (programmerad celldöd) endast av cancerceller men inte av friska celler från däggdjur. (Figuren visas inte här med kan återfinnas i originalartikeln som figur 5d.)
Ökad syntes av p53 – som ökar med ökad dos av Fenbendazol.
Man kunde också visa att Fenbendazol framkallade ökad syntes av p53, vilket visar att det är aktivt i cellkärnan och påverkar DNA:t och genen TP53 till att producera mera p53. Denna ackumulering av p53 i cellkärnan är dosberoende – och ju högre dos Fenbendazol desto större syntes av p53 (se fig. 5). En tidigare studie hade dessutom visat att Fenbendazol framkallar mitokondriell translokation av p53. [Marchenko ND, Wolff S, Erster S, Becker K, Moll UM. Monoubiquitylation promotes mitochondrial p53 translocation. EMBO J. 2007;26:923–934]. Detta korrelerar väl med ökad apoptos av cancerceller när Fenbendazol ges.
Bakgrund till mekanism 3:
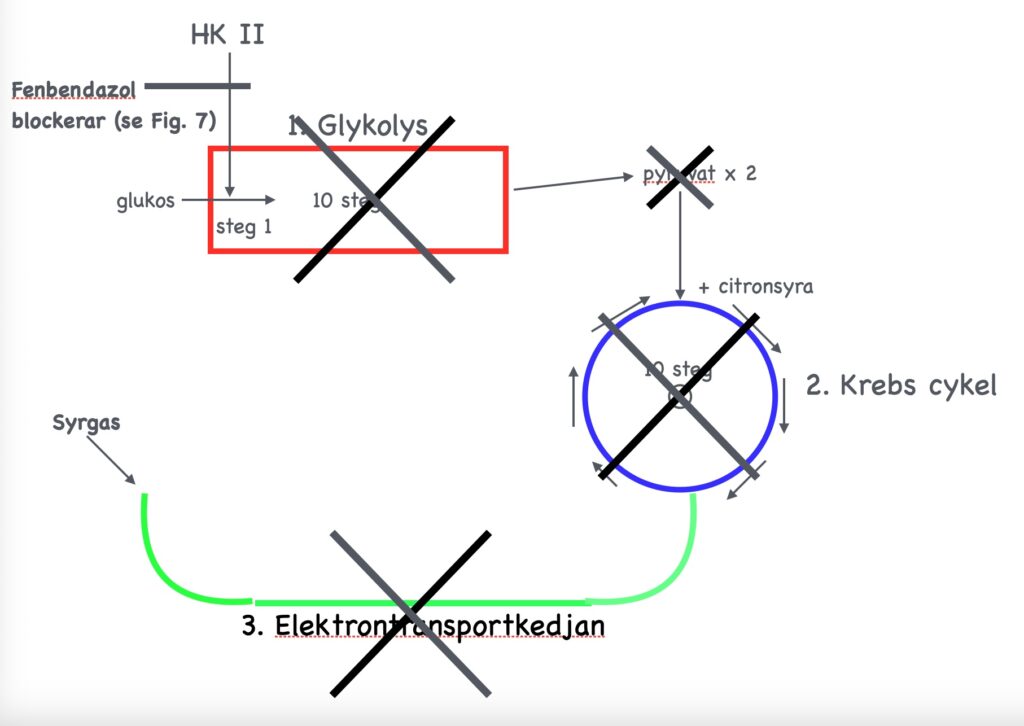
Omsättningen av glukos för att skapa energi i en normal frisk cell sker i tre steg;
1. glykolys (i stort sett sönderdelning av sockermolekylen i två lika stora delar)
2. citronsyra-cykeln och
3. elektrontransport-kedjan.
Om allt detta kan ske så bildas det totalt 38 små energipaket (ATP, adenosintrifosfat) av en enda sockermolekyl (glukos), varav 2/38 energipaket bildas under det första steget, glykolysen och de andra 36/38 under de två andra. Detta under förutsättning att det finns tillräckligt med syrgas i blodet. Om det inte finns tillräckligt med syrgas så kommer inte de två sista stegen att kunna ske utan det bildas då endast 2 ATP samt en stor mängd mjölksyra, vilket är välkänt för dem, som utför utmattande idrottsprestationer.
Det har emellertid varit känt sedan c:a ett hundra år (den s.k. Otto Warburg-effekten, Nobelpriset 1931) att cancerceller, trots full tillgång till syrgas, bara kan utnyttja det första steget, glykolysen, och av en sockermolekyl endast skapa 2 istället för 38 små energipaket (ATP). Cancercellerna kan bara tillgodogöra sig sockret på anaerob väg (=utan syrgas) vilket gör att dessa celler går miste om 95% sockrets energiinnehåll. Det är därför som cancerpatienter magrar kraftigt. Eftersom cancercellerna kräver nästa 20 gånger så mycket socker som friska celler för att skapa samma mängd energi, så stjäl cancern näringen från övriga friska celler.
Cancercellens upptag av socker blockeras
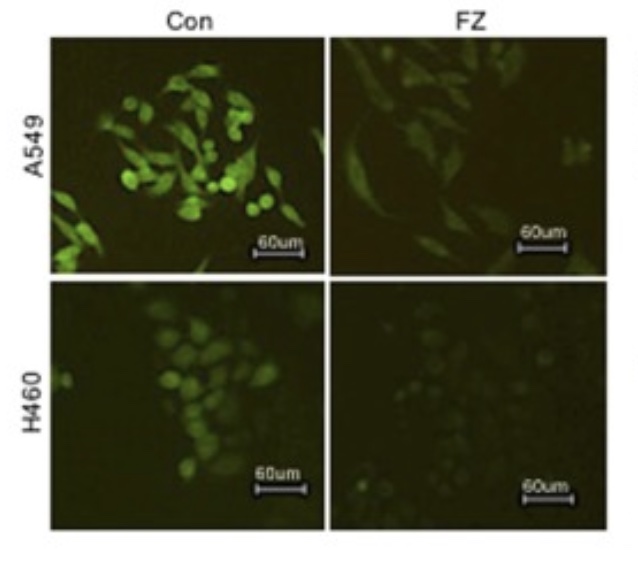
Det har nu visat sig att Fenbendazol hämmar cancercellens upptag av glukos på flera olika sätt vilket leder till att cancercellerna genomgår apoptos. I figur 6 nedan har man en använt ett fluorescerande spårämne som används för att övervaka glukosupptag i levande celler, kallad 2-NBDG. I de båda vänstra kontrollgrupperna (Con) kan ses hur glukos upptas i mycket stor utsträckning (lyser grönt) i båda cellinjerna (icke-småcellig lungcancer från cancerpatienter). Till samma cellinjer har man till försökscellerna FZ givit 1µM Fenbendazol under 4 timmar innan dessa fick glukosanalogen 2-NBDG. Man ser då att glukosanalogen knappast upptas alls i de cancerceller som fått FZ, vilket är ett bevis på att Fenbendazol blockerar upptaget av socker och svälter ut cancercellerna.
Blockeringen är dosberoende
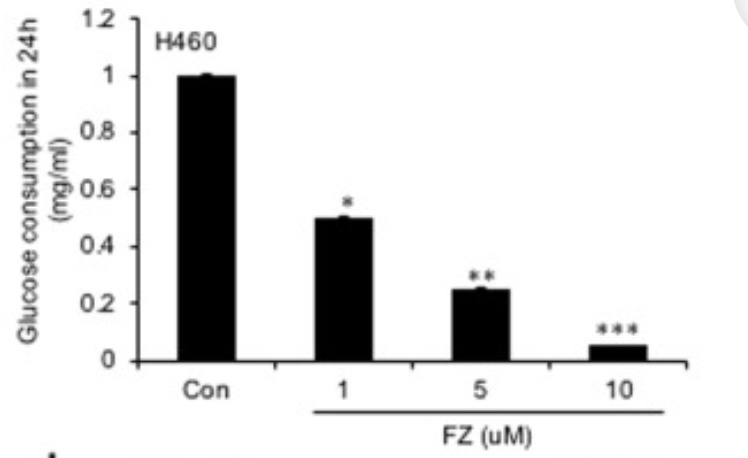
I ett annat försök på H460-celler (icke-småcellig lungcancer) gav man ökande doser av Fenbendazol (FZ) under 24 timmar och mätte sedan hur mycket glukos som konsumerats beroende på hur stor dos som givits. Man kan se att med 1 µM, 5 µM och 10µM Fenbendazol så minskar glukoskonsumtionen beroende på dosens storlek med respektive 50%, 75% och 95%. Stjärnorna ovanför staplarna visar på statistiskt signifikanta minskningar (*p<.05, **p<.01, ***p<.001).
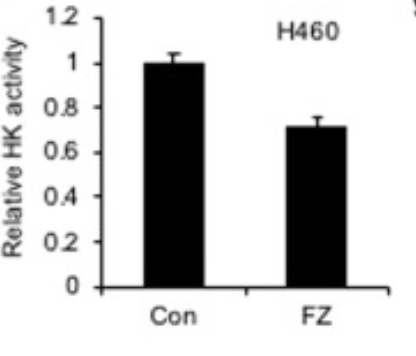
Hexokinas II – ett nyckelenzym till glykolysen
När glukos kommer in i en cell så kopplas inom mikrosekunder en fosfatjon på glukosmolekylen med hjälp av ett enzym, Hexokinas II, varvid glukos omvandlas till glukos-6-fosfat, som är en förutsättning för glykolysen. Hexokinas II är därför ett nyckelenzym. I detta försök undersöktes hur Fenbendazol påverkade Hexokinas II:s aktivitet – ökade det eller minskade det dess aktivitet? Som vi ser på grafen så minskade det dess aktivitet med c:a 30%, vilket således är en av förklaringarna till att cancercellens konsumtion av socker minskar. I detta fall undersökte man dock bara effekten av 1 µM och inte ökande doser av5µM och 10 µM som i studien ovan. Det är dock högst rimligt att förmoda att ökande doser Fenbendazole skulle minska dess aktivitet ytterligare.
Citronsyrecykeln
Nästa steg,Citronsyrecykeln (Kreb’s cycle), som utspelas i mitokondrierna, innehåller förberedelser för att energi skall utvinnas ur det socker som genomgått glykolys och omvandlats till två halva sockermolekyler, pyruvat). Krebs cykeln innehåller 8 olika steg och för varje steg får molekylen ett nytt namn. Det enzym, som omvandlar det 6:e steget (succinat) till det 7:e (fumarat) heter succinatdehydrogenas (SDH).
Enzymet SDH motverkar utveckling av cancer
Förutom att vara ett viktigt enzym i citronsyrecykeln är det känt att SDH i sig själv motverkar tumörbildning. Man har känt till att det funnits ett samband mellan mutationer i detta enzym och binjuremärgstumörer, andra neuroendokrina tumörer och njurcancer. Man har också känt till att det funnits ett samband mellan en minskad mängd SDH och mag- och tjocktarmscancer. Men man har inte känt till mekanismen bakom detta samband.
I en studie kunde man visa att ansamlingen av succinat i cellvätskan (cytosolen) utanför mitokondrier, som en följd av minskad insöndring av SDH, signalerade till ett annat enzym att utveckla cancer. [Selak MA, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 2005;7:77–85.] Det är därför viktigt att hög aktivitet av SDH upprätthålls. Om citronsyrecykeln stannar upp vid succinat och inte går vidare till fumarat så ansamlas succinat, som kan göra att cancer utvecklas. [Som en intressant parentes kan nämnas att enzymet fumaratreduktas, som bara finns hos parasiter, går precis motsatt väg och hos mikrober och lägre organismer omvandlar fumarat till succinat. Eftersom det finns röster i cancerdebatten som hävdar att cancer orsakas av intracellulära parasiter, så kan detta vara värt en tanke. Detta skulle då eventuellt kunna förklara Nobelpristagaren Otto Warburgs tes från 1920-talet att cancerns gåta är att mitokondrierna inte fungerar.]
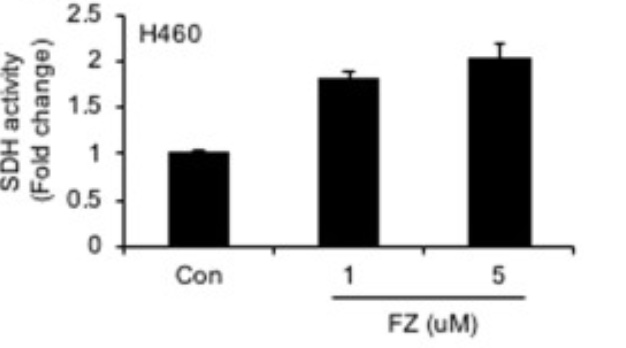
Fenbendazol ökar aktiviteten av SDH
Fenbendazol spelar här en positiv roll. Vi ser nedan i fig. 9 hur aktiviteten av SDH i H460-cancercellen ökar med 75 – 100 % om 1 respektive 5 µM av Fenbendazol (FZ) tillförs cancercellerna.
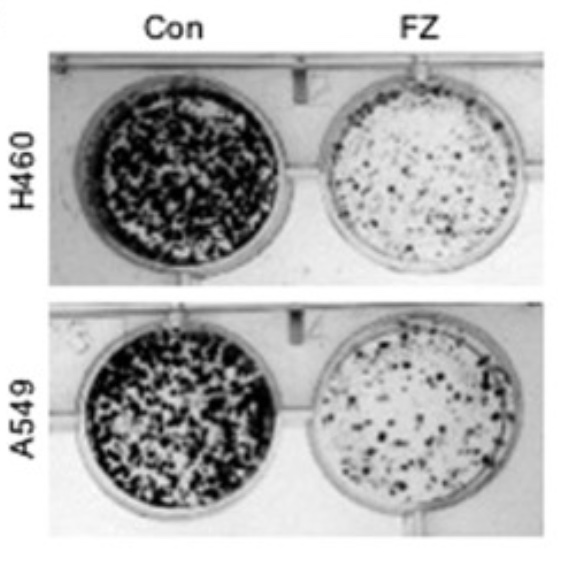
FZ hämmar effektivt kolonibildning av humana icke småcellig lungcancer-celler i odlingskultur.
Behandling av A549- och H460-celler med 1 uM Fenbendazol (FZ) under 48 timmar resulterade i en signifikant minskning av antalet kolonier jämfört med obehandlade kontrollceller (Fig. 10).
Man har också försökt att kvantifiera tumörtillväxten under antal dagar vilken kurva ses nedan i Fig. 11.
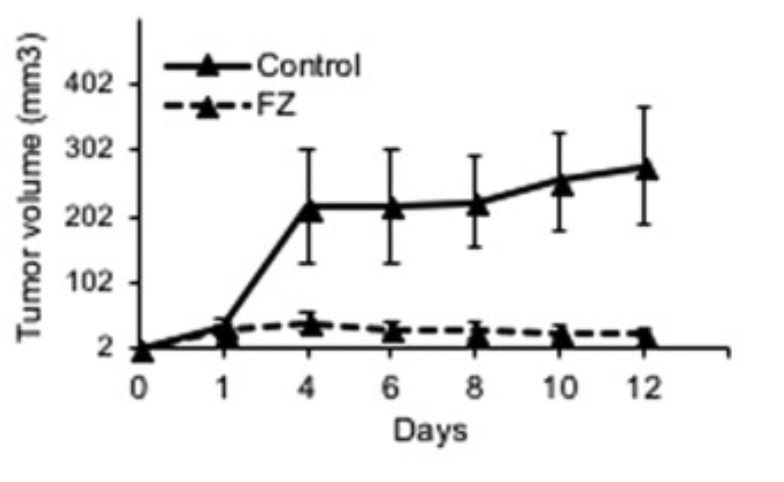
Tydlig effekt även in vivo
Detta tyder på att Fenbendazol (FZ) är ett potentiellt anti-neoplastiskt medel som dödar cancerceller inte bara in vitro utan också in vivo. Dessa data stämmer väl överens med in vitro-analysen av FZ-medierad celldöd.
Förstärker cytostatikas effekt
Nu återstår frågan hur detta går ihop med cytostatika. Det visade sig att taxol, som är ett cytstatikum, fick synergistisk (förstärkt) effekt på cancer när det kombinerades med Fenbendazol. Så man riskerar ingenting när man kombinerar Fenbendazol med cytostatika. Tvärtom. [Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55.]
Tillägg: Förklarande bilder
Fig. 13.
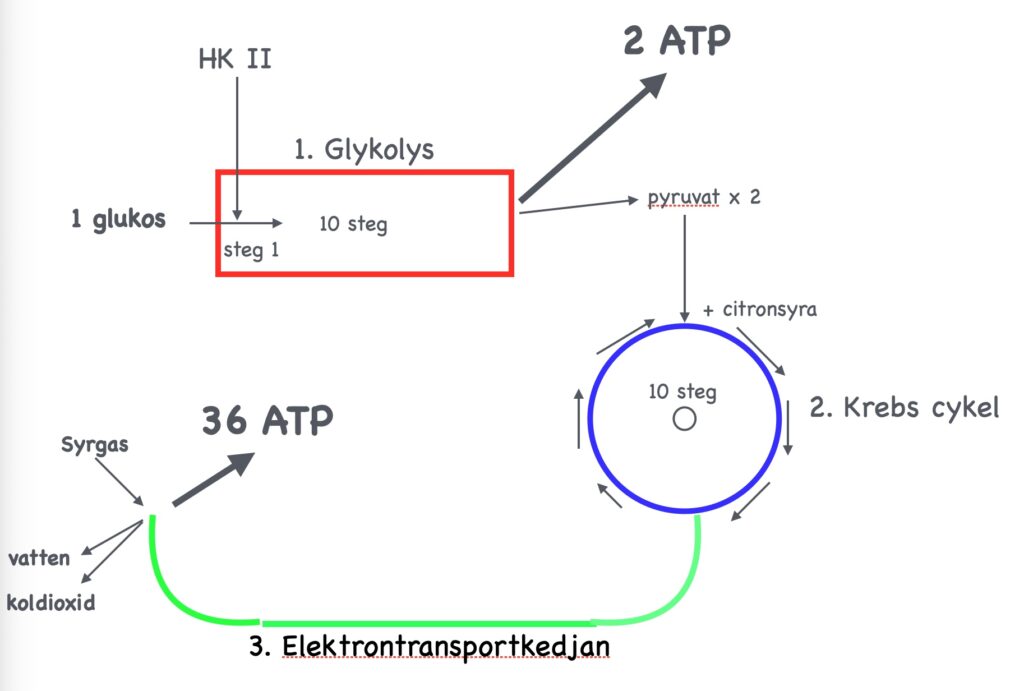
Citronsyra-cykeln kan liknas vid ett vattenhjul som alstrar energi (Tänk England i industrialismens barndom). Det börjar högst upp (kl. 12) med citronsyra som förenklat uttryckt fylls på med en pyruvatmolekyl vilket sätter fart på hjulet. Denna kombination, pyruvat+citronsyra, genomgår därefter 10 olika steg varigenom det bildas två olika typer av energialstrande molekyler samtidigt som pyruvatet förbrukas. Slutresultatet när hjulet snurrat ett varv blir återigen citronsyra (ungefär som när klockan börjar om på kl. 1 när det gått 12 timmar). Och så sätter samma procedur igång igen som en evighetsmaskin. De energialstrande molekylerna går sedan vidare till elektrontransport-kedjan där de 36 energipaketen (ATP, adenosintrifosfat) bildas. [Det bör observeras att de två sista processerna, citronsyra-cykeln och elektrontransport-kedjan äger rum i cellens energiverk, mitokondrierna, av vilka det kan finnas flera tusen i en enda cell. Glykolysen däremot äger inte rum i mitokondrierna utan fritt i cellens cytoplasma.]
Fig. 14.
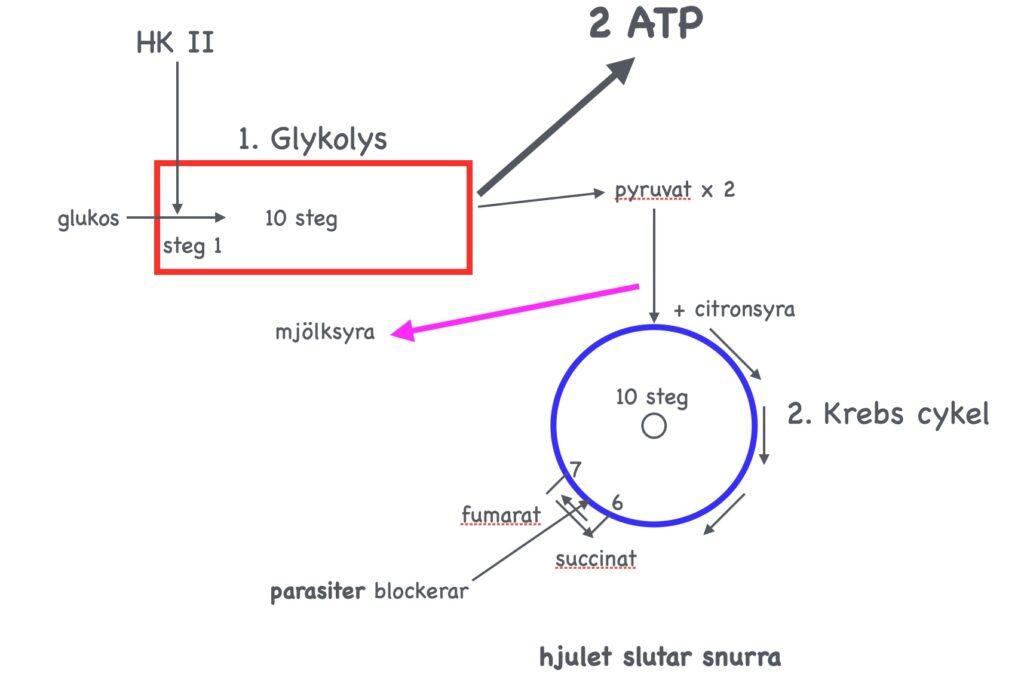
Fig. 15.
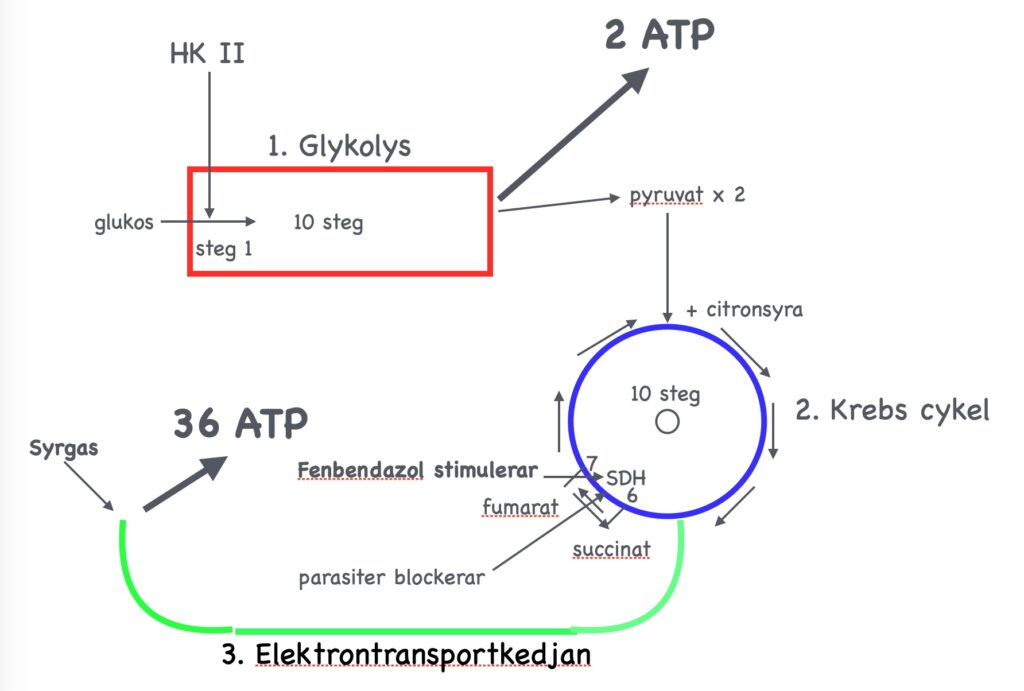
Fig. 16.